Image credit: Dr. Fubiao Shi
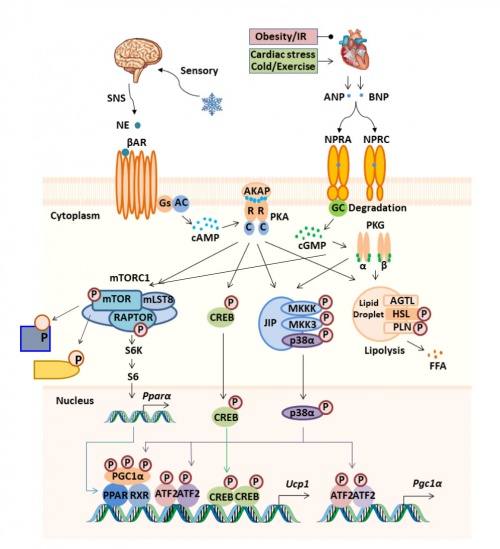
Our research interests started with the beta-adrenergic receptors in the adipocyte, since all three (β1AR, β2AR, β3AR) are present to varying degrees and depending upon species. The βARs regulate adipocyte metabolism and energy expenditure in brown adipocytes by mitochondrial non-shivering thermogenesis, through activating production of the second messenger cAMP and subsequent downstream signaling pathways, a number of which our lab discovered. These findings, which have eventually become major efforts were the result of ‘accidents’ or ‘strange’ observations. This proved to be the case for identifying the p38 MAP kinase as a downstream mediator of PKA to activate the transcription of the uncoupling protein-1 (UCP1) and Ppargc1a (PGC1α) genes (Cao et al. JBC 2001; Cao et al. Mol Cell Biol 2004; Robidoux et al. Mol Cell Biol 2005), as well as for how the cardiac natriuretic peptides (NPs) ANP and BNP and their second messenger cGMP promote a parallel pathway to the catecholamines and cAMP to drive adipose ‘browning’ and energy expenditure (Bordicchia et al. J Clin Invest 2012). It has been known for some time that obesity in animal models as well as humans in accompanied by an increase in the level of NP receptor NPRC, also called the ‘clearance receptor’ because of its ability to bind, internalize and degrade the NPs. The use of various genetically engineered mouse models illustrates that removing NPRC from the adipocyte allows extended NP signaling, protecting from obesity and insulin resistance (Bordicchia et al. J Clin Invest 2012; Wu, Shi et al. Sci Signaling 2017). The work on NPRC continues to be active in the lab. We also recently reported another mechanism by which NPRC can interfere with NP signaling in cells – by forming non-functional heterodimers with the signaling receptors NPRA and NPRB (Liu et al. Proc Natl Acad Sci 2023). We are also examining how the Nprc gene itself is regulated (Shi et al. J Biol Chem 2021), as well as how other components of the signaling system, include the cGMP phosphodiesterase-9 (Pde9) contributes to signaling strength (Ceddia et al. Diabetes 2021).
The most unexpected ‘accidental’ finding that has turned out to be another major topic in the lab was that βAR agonists could activate the rapamycin-sensitive mTOR complex (mTORC1) and that this process appears necessary to both promote ‘browning’ of white adipose tissue in response to cold or selective β3AR agonists, as well as the proper development of the interscapular brown fat (Liu et al. J Clin Invest 2016). The mechanism is, at least in part, driven by the direct phosphorylation of mTOR and Raptor by PKA (and we now know that the natriuretic peptides and cGMP do the same thing, through the cGMP-dependent protein kinase (PKG) (Liu et al. Mol Metab 2018). We began searching for substrates of PKA-activated mTORC1 with our collaborator Dr. Marcus Krüger, two of which we have studied in some detail (Crowder et al. Physiol Reports 2023; Shi et al. Mol Metab 2023). We have now generated genetically modified mice in which a point mutation is introduced into the Raptor sequence that removes the Serine residue that is phosphorylated by PKA Ser791, replacing it with either an Alanine residue, which cannot be phosphorylated – or with an Aspartic Acid residue, which creates a phospho-mimetic residue. These mutations have been engineered in the adipocytes of mice as well as in some other cell types with our collaborators. Preliminary studies in these mice show clear metabolic phenotypes, reinforcing the importance of this cyclic-nucleotide activated mTORC1 pathway in physiology.
Our funding sources include the NIH as well as other foundation support.
https://reporter.nih.gov/search/QrS9s8BzTk2jFmhQ9T90Ag/projects