Gannon Lab
The primary area of research in the Gannon Lab at Vanderbilt University Medical Center, led by PI Dr. Maureen Gannon, is molecular and cell biology of pancreas development, function and regeneration.
The pancreas is essential for normal digestion and maintenance of blood sugar levels. It is composed of an exocrine compartment, made up of acinar and ductal cells that secrete and transport digestive enzymes, as well as an endocrine compartment (the Islets of Langerhans), made up of alpha cells that produce glucagon, beta cells that produce insulin, delta cells that produce somatostatin, and PP cells that produce pancreatic polypeptide.
We study the role of genes and signaling pathways involved in the development, function, and regeneration of specific cell types within the pancreas.
Current research projects in the Gannon Lab
Effects of in utero environment on β-cell development and function
Lexi DelBene, Graduate Student
The focus of this project is identifying the role of oxidative stress pathways in the development and maintenance of functional beta cell mass.
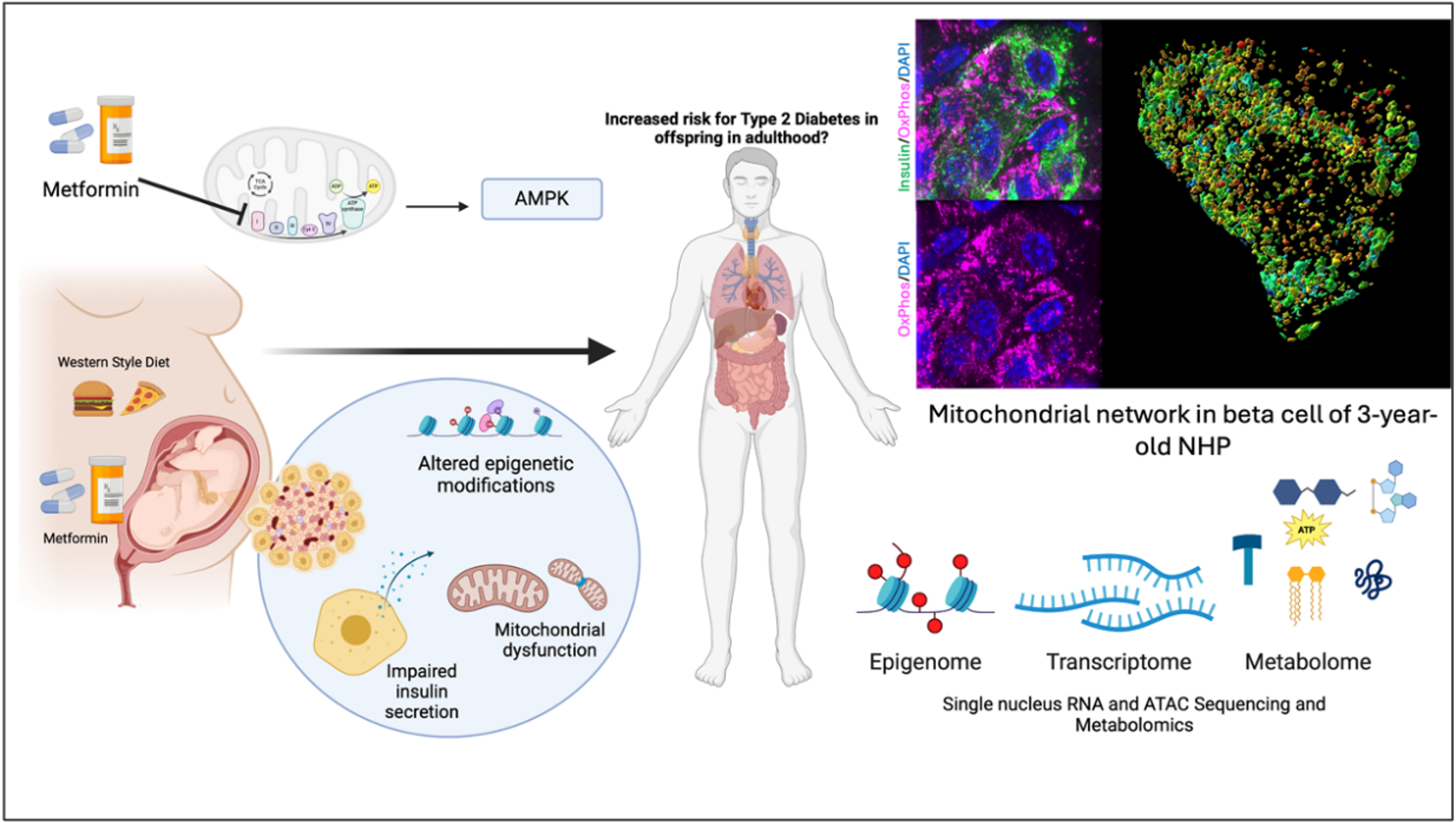
One aspect of this project examines the consequence of changes to antioxidant stress in the presence of metformin, a common Type 2 Diabetes therapeutic, on NHP offspring islet function during fetal development and as they approach adolescence.
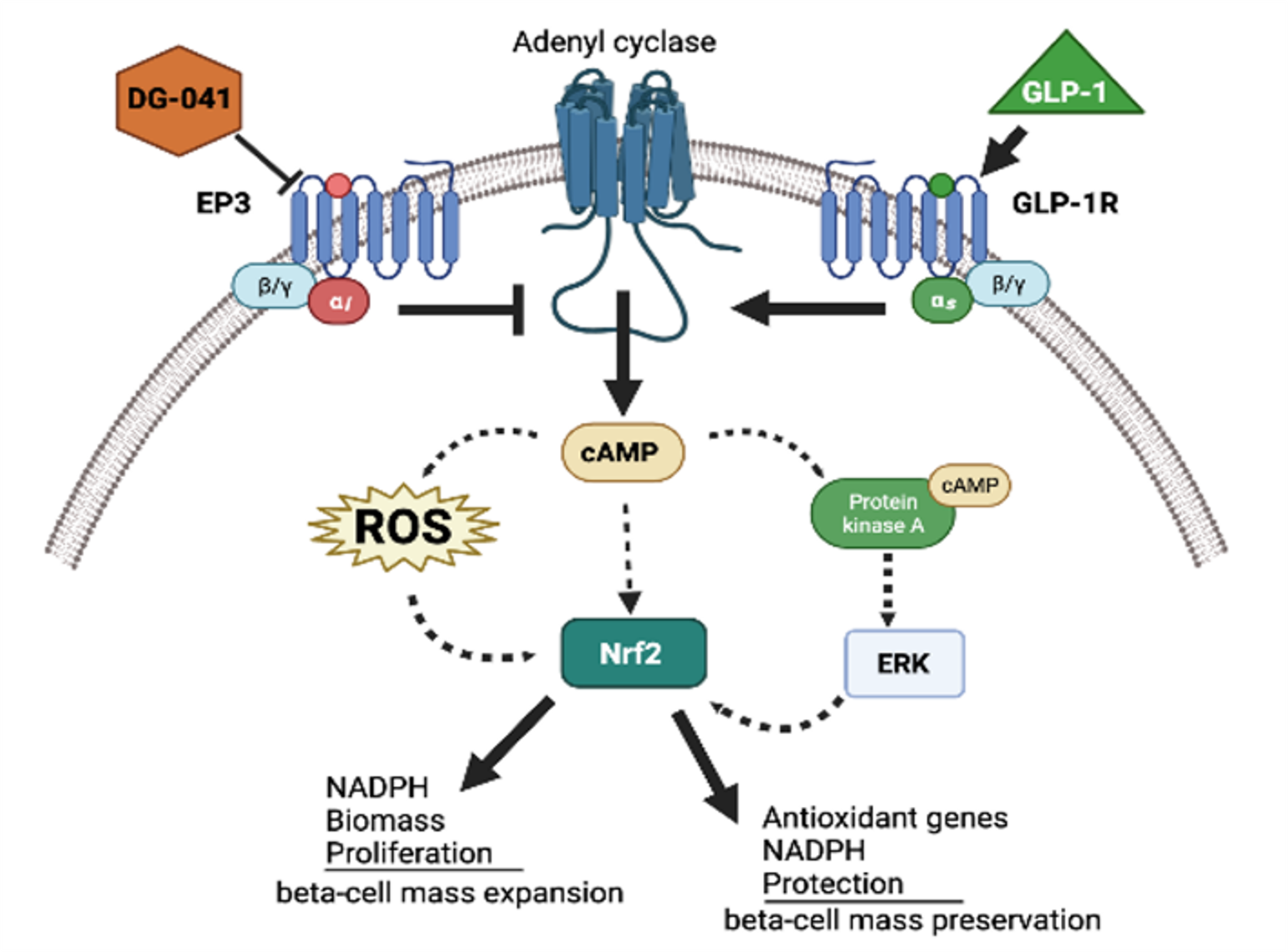
The other aspect of my project builds on previously published results in a mouse model of T2D where antioxidant stress in adult islets is ameliorated by treatment with an inhibitor of the GPCR, EP3, whose ligand, PGE2 is a metabolite of arachidonic acid. EP3 antagonism was also demonstrated to increase the expression of the GLP-1R in beta cells, identifying the interplay between these pathways may provide insight into how combined T2D therapeutics can be utilized to improve human health.
Signaling pathways that enhance adult β-cell proliferation and survival
Micaela Maxwell, Graduate Student
Elevated levels of oxidative stress are one of the leading causes for the development of diabetes mellitus. Nrf2 is a redox-sensitive, basic leucine zipper transcriptional factor that upregulates antioxidant gene expression by binding to the promoter region of the antioxidant response elements (AREs).
It has been shown that on chronic hyperglycemia β-cell identity gene expression, insulin content, and β-cell mass, resulting in decrease glucose tolerance. Nrf2, once activated, dissociates from Keap1 in the cytoplasm and translocates into the nucleus which then binds to AREs signaling antioxidative genes, NADPH production, nucleic acid synthesis, and phospholipid syntheses.
Therefore, Nrf2 has great potential therapeutic value for treating the β-cell failure in diabetes. We are investigating the activation or overexpression of Nrf2 to try and increase proliferation and β-cell mass in diabetic mouse models.
Re-educating immune cells to prevent β-cell stress and death
Julie Burkett, PhD candidate
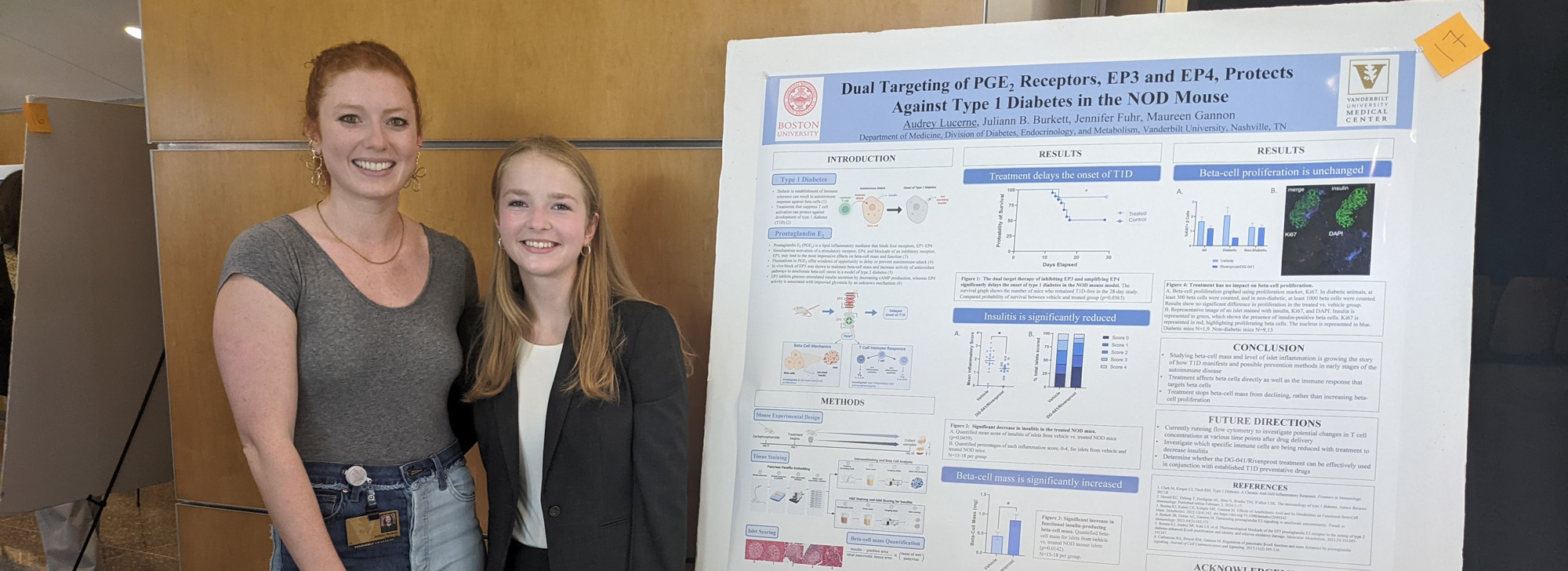
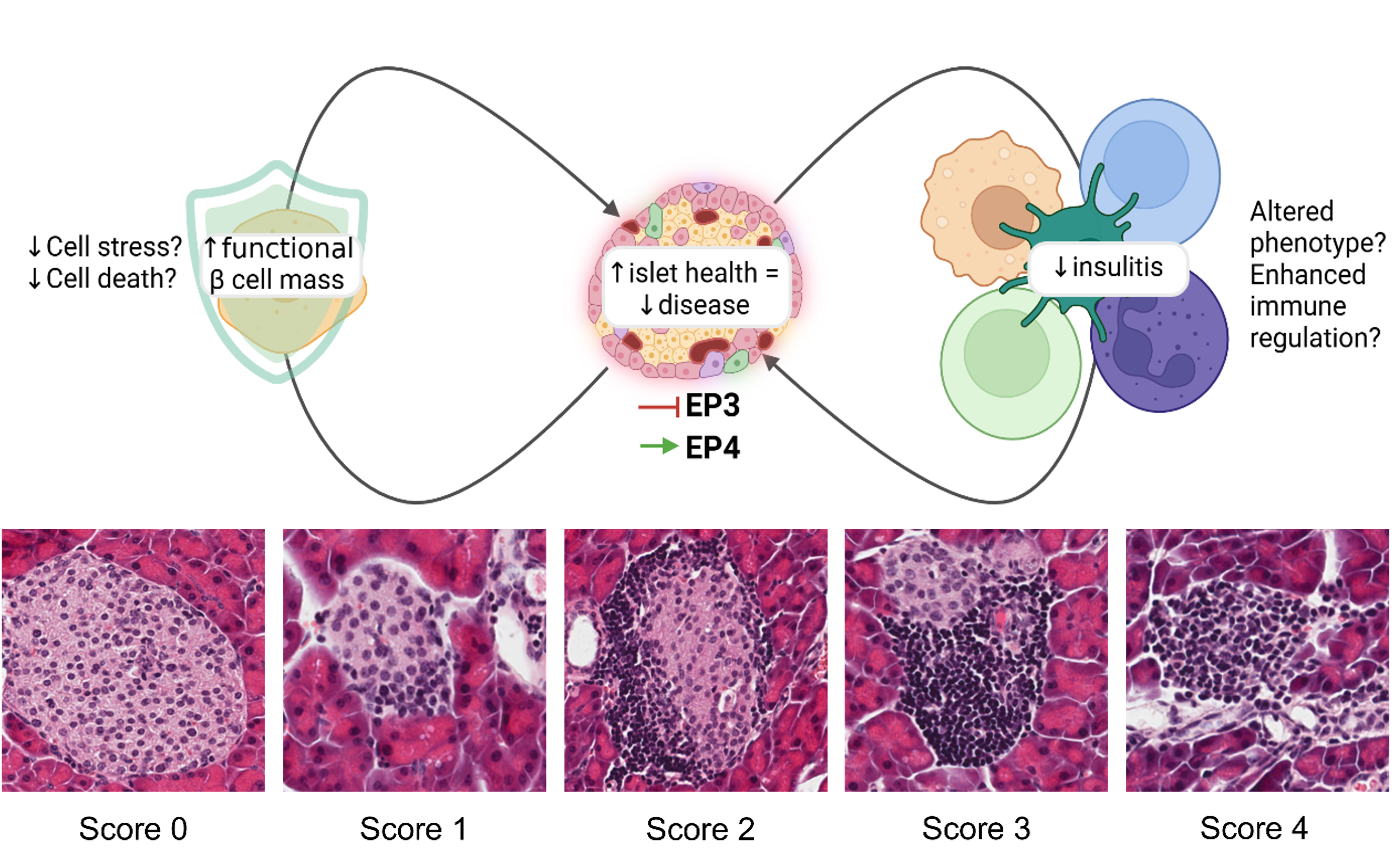
Type one diabetes (T1D) is a chronic autoimmune condition resulting from the progressive destruction of insulin-producing β cells, leading to irreversible dysglycemia. It is well accepted that both immune system and β-cell dysfunction contribute to T1D pathogenesis, however many investigations of interventions focus on either the β cell or the immune system. Manipulation of the PGE2 signaling pathway, through its functionally opposing receptors EP3 and EP4, may provide an intervention which can simultaneously alter the autoimmune attack and provide cell-autonomous protection to the β cells, allowing delay, prevention, or reversal of disease pathogenesis.
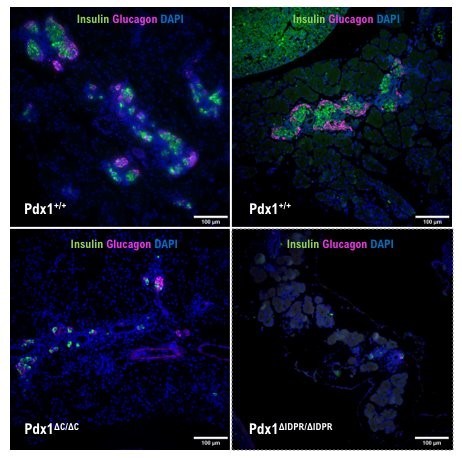
Interactions of Pdx1 and Oc1 transcription factors to promote β-cell differentiation
Anthony Wokasch, PhD candidate
My dissertation research in the Gannon lab focuses on the role of the Pdx1 IDPR in:
- Endocrine specification and differentiation during embryogenesis
- Regulation of Pdx1 protein localization and levels within beta cells. It has been shown that the C-terminus of Pdx1 is required for proper endocrine development.
My work specifically focuses on how removal of just the IDPR within the terminus of Pdx1 effects pancreatic endocrine specification and differentiation in vivo. I collect mouse embryos at various stages of pancreas development and use a combination of immunohistochemical techniques to evaluate markers for specific cell lineages.
Additionally, the Gannon lab has shown that Pdx1 protein localization and levels change throughout the cell cycle. Therefore, I am expressing mCherry-tagged constructs of Pdx1 protein variants in immortalized INS-1E beta cells to determine how manipulating the C-terminal tail affects Pdx1 protein localization and localization of its interaction partners, Oc1 and SPOP.
Life in the Gannon Lab