Research
How does mitochondrial dysfunction alter innate immune outcomes and confer susceptibility to infection?
Mitochondria are central players in the innate immune response that play a key role in regulating cell-intrinsic responses to Mtb infection. Because many pathways activated by mitochondrial damage-associated molecular patterns are crucial for controlling Mtb infection (e.g., cell death, type I IFN induction, selective autophagy), there is a critical need to elucidate molecular mechanisms that drive their regulation. Our group seeks to uncover how danger signals from mitochondria and bacteria are integrated to elicit innate immune responses, providing crucial insights into the molecular mechanisms that drive genetic susceptibility to mycobacterial infection with the hope of identifying new targets for host-directed therapeutic intervention.
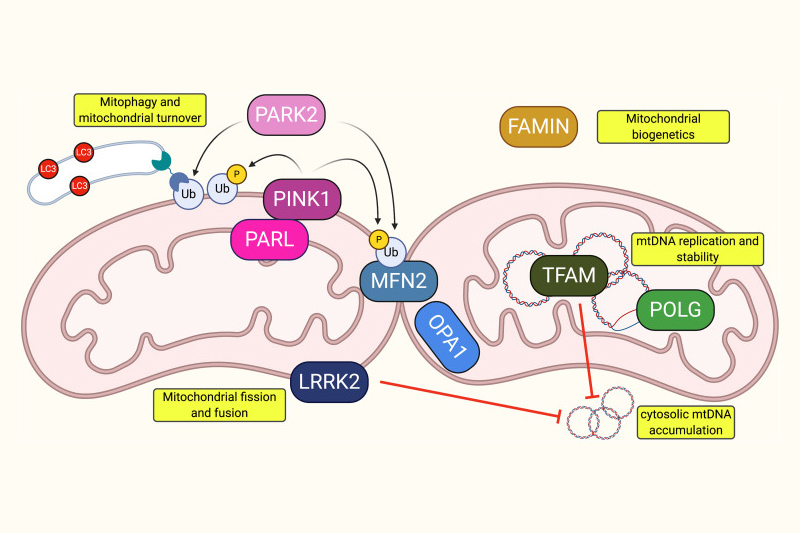
Mutations in functionally diverse mitochondrial proteins confer susceptibility to mycobacterial disease. Shown is a representation of proteins associated with leprosy/tuberculosis susceptibility via either GWAS or direct experimental investigation. From left to right, proteins are Presenilins-associated rhomboid-like protein (PARL), Parkinson’s disease 2 (PARK2), PTEN-induced kinase 1 (PINK1), leucine-rich repeat kinase 2 (LRRK2), mitofusin 2 (MFN2), optic atrophy 1 (OPA1), fatty acid metabolism-immunity nexus (FAMIN), mitochondrial transcription factor A (TFAM), and mitochondrial DNA polymerase gamma (POLG). https://doi.org/10.1128%2FIAI.00687-20
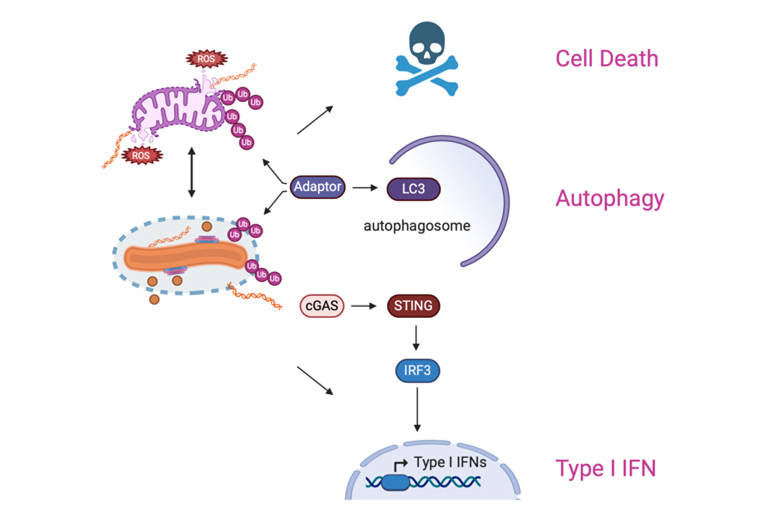
Schematic of the innate immune pathways that are activated by both Mtb and mitochondrion-derived PAMPs and DAMPs. Our goal is to identify both host and bacterial factors that modulate mitochondrial homeostasis and tip the balance between protective versus pathogenic responses.
What are the cellular triggers of hyperinflammatory cell death? Can we identify intervention points to develop novel therapies?
The question of why some tuberculosis patients develop acute/severe disease while most do not remains one of the most pressing in the field. We propose that the spectrum of TB disease is strongly influenced by the nature of inflammatory cell death pathways engaged by macrophages in response to Mycobacterium tuberculosis (Mtb) infection.
We recently identified a new type of cell death that preferentially occurs in macrophages experiencing mitochondrial dysfunction and oxidative stress. This new flavor of cell death, dubbed GSDMD-mediated necroptosis, preferentially occurs in macrophages subjected to oxidative stress. We have linked this new type of cell death to excessive neutrophil infiltration and hyperinflammation in a mouse model of tuberculosis infection. Building upon this work, we seek to identify novel drivers of inflammatory cell death in macrophages and define how oxidative stress disrupts the balance of protective versus pathogenic cell death modalities during Mtb infection. Our ultimate goal is to identify novel intervention points that may be more efficacious to target in TB patients with underlying genetic factors, coinfections, or comorbidities associated with oxidative stress.
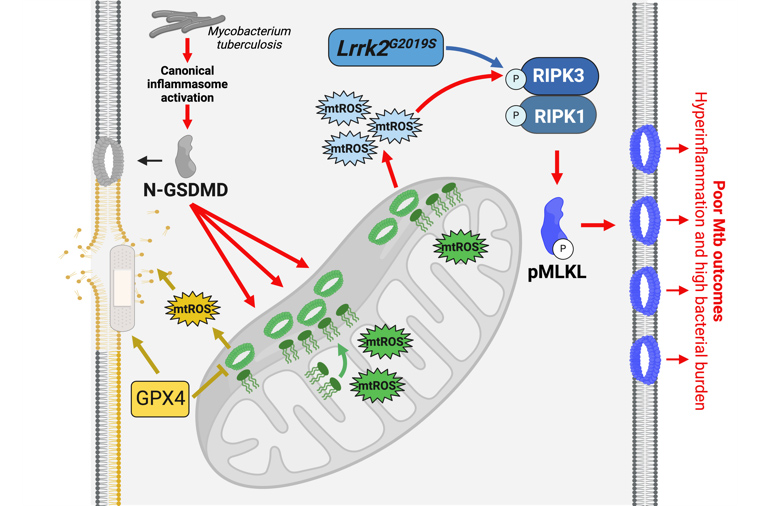
Schematic representation of GSDMD-mediated necroptosis (red arrows) during Mtb infection of macrophages. (blue) will test if LRRK2 acts as a bona fide pro-necroptosis kinase; (green) will identify novel triggers of GSDMD relocalization to mitochondrial membranes; and (yellow) will investigate links between lipid peroxidation, the glutathione peroxidase GPX4, and GSDMD-mediated necroptosis.
How do macrophages regulate innate immune signaling to combat infection?
For macrophages to mount a strong—yet measured—response to pathogens, many immune proteins need to be dynamically regulated—i.e. turned on or off by subcellular relocalization, altered binding partners, post-translational modification, etc. Despite their importance in balancing immune responses, the signals and factors that orchestrate this functional diversification—and thus help cells mount a balanced response to pathogens—remain poorly defined. We are currently elucidating how a newly described TRIM14-TBK1-STAT3 axis dictates innate outcomes in response to a variety of immune stimuli, including infection with Mycobacterium tuberculosis in macrophages ex vivo and in vivo, leveraging our Mtb mouse model. This work will make fundamental insights into the molecular mechanisms through which TRIM14 functionalizes TBK1 and STAT3, while also assessing the viability of targeting this axis to help ameliorate immunopathology in diseases like tuberculosis. This signaling axis is also important beyond the context of Mtb infection, as defects in TBK1 are associated with diverse and debilitating human diseases such as ALS, systemic autoinflammation, and certain cancers.
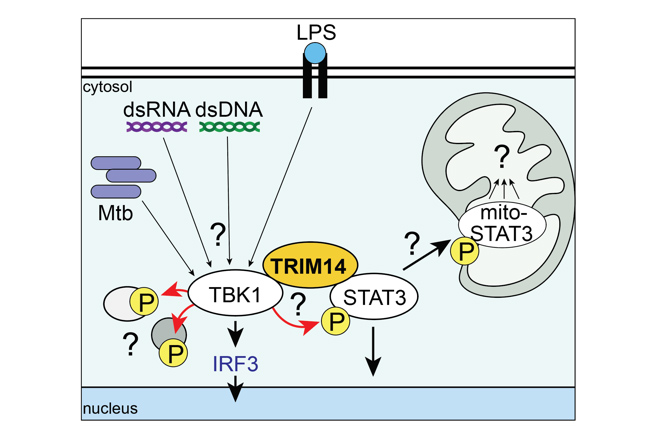
Schematic of the TRIM14-TBK1-STAT3 axis. Gaps in knowledge include: How do different signals control TRIM14/TBK1/STAT3 interactions? How is TBK1 substrate choice directed by TRIM14? How does phosphorylation target STAT3 to mitochondria and what does it do there?
New Projects
Many of the genes we study, such as LRRK2, are associated with chronic diseases like Parkinson's and Crohn's disease, as well as susceptibility to other infections. Therefore, we aim to study how metabolism shapes innate immune responses in disease models beyond M. tuberculosis. With this in mind, we are building on our expertise and have several new projects in the lab. First, we want to understand whether altered mitochondrial function impacts inflammation at other sites.