History and Impact
Interstitial Lung Disease Center
The Interstitial Lung Disease Clinic at Vanderbilt was founded in the late 1990s by Dr. James Loyd and grew into the Interstitial Lung Disease Center under the leadership of Dr. Lisa Lancaster. The ILD Center at Vanderbilt has cared for thousands of patients with idiopathic pulmonary fibrosis (IPF) and other interstitial lung diseases over the past 25 years.
The center was a charter member of the Pulmonary Fibrosis Foundation Care Center Network and has been highly engaged in clinical and translational ILD research and IPF/ILD clinical trials.

IPF/ILD clinical trials
Since its inception, the Interstitial Lung Disease Center at Vanderbilt has been one of the leading IPF/ILD clinical trials centers in the country. Vanderbilt was a site for the very first IPF clinical trial, served as one of the NIH-supported IPFNet study sites, and was a top enrolling site for the pirfenidone and nintedanib trials that led to the first FDA-approved treatments for IPF and progressive pulmonary fibrosis (PPF).
The clinical trials program is continuously enrolling in ongoing studies, and information about currently active studies is available at the ILD center.
Familial pulmonary fibrosis (FPF)
The Vanderbilt Interstitial Lung Disease Program has long been a leader in the study of familial pulmonary fibrosis. Since its founding in the early 2000s, the Familial Pulmonary Fibrosis registry has enrolled pulmonary fibrosis patients from more than 790 families and includes thousands of individuals.
Studies from the FPF registry have been instrumental in identifying the genetic basis of Familial Pulmonary Fibrosis. In 2002, studies of FPF at Vanderbilt led to the discovery of the first genetic cause of FPF (mutations in SFTPC, the gene encoding for surfactant protein C). In 2007, another study of families from the Vanderbilt FPF registry (in collaboration with Dr. Mary Armanios and colleagues at Johns Hopkins) discovered that telomerase mutations are a cause of FPF.
Over the past 15 years, studies of families in the FPF registry have led to the identification of the MUC5B risk genotype, discovered numerous other FPF risk genes, and established the frequency different gene mutations are found in FPF families. This work led to the first clinical guidance for genetic testing in FPF.
Familial pulmonary fibrosis research at Vanderbilt has been heavily focused on understanding how to identify and care for relatives of FPF patients. Since 2008, supported by the NIH/NHLBI, Vanderbilt has led an ongoing, prospective study of FPF relatives. Started by Drs. Jim Loyd, Timothy Blackwell and William Lawson, and now led by Drs. Margaret Salisbury and Jonathan Kropski, this study includes more than 500 FPF relatives and has been instrumental in establishing the natural history of FPF.
Key findings from these studies include identifying the earliest signs of FPF that can be detected on CT scans, determining genetic and environmental factors that influence the development of FPF, and characterizing the progression risk of FPF relatives with early signs of pulmonary fibrosis.
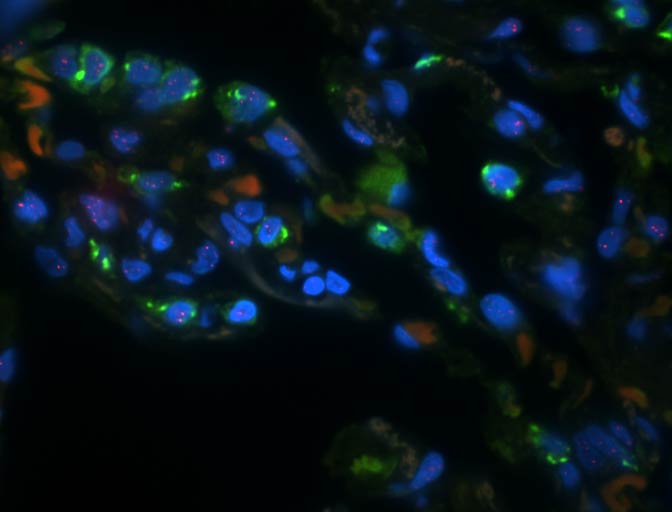
Short telomeres in type 2 alveolar epithelial cells precede the development of interstitial lung abnormalities in FPF relatives. Learn more about this research here.
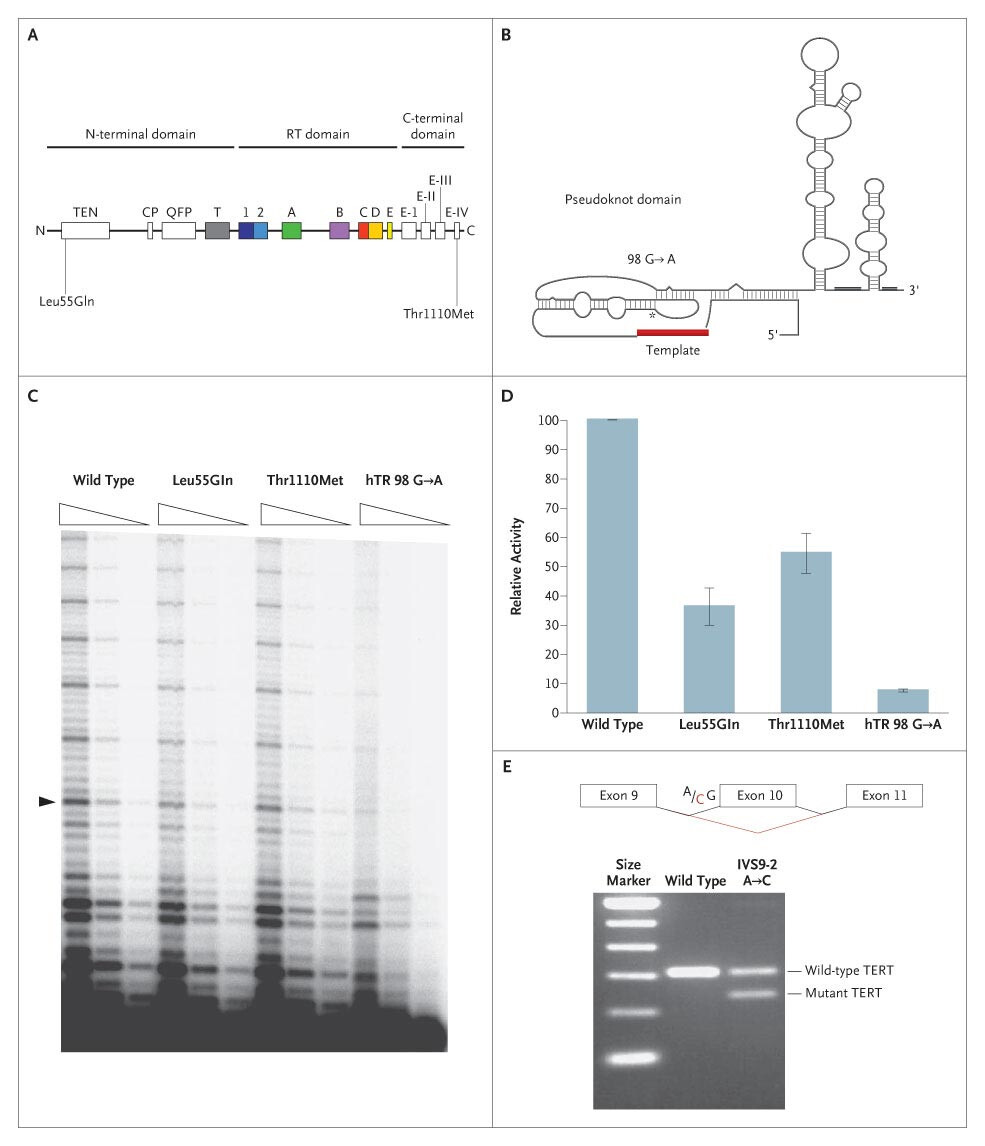
The chart shows how familial pulmonary fibrosis-associated telomerase mutations leads to reduced telomerase activity and telomere shortening.
Learn more about "Telomerase mutations in families with idiopathic pulmonary fibrosis."
Pulmonary fibrosis biology
Complementing the clinical trials and clinical research in the ILD Center, Vanderbilt has been home to a robust basic and translational research program that has made key discoveries into the mechanisms that contribute to our current understanding of the molecular pathogenesis of pulmonary fibrosis.
The discovery of SFTPC mutations as a cause of FPF was a highly impactful finding that provided the first and most direct evidence that dysfunction of the lung epithelium is central to PF biology. Subsequent studies revealed that SFTPC mutations (and several other factors) lead to endoplasmic reticulum stress and activation of the unfolded protein response in the IPF lung epithelium, a near universal finding that contributes to dysfunctional repair after injury to the lung.
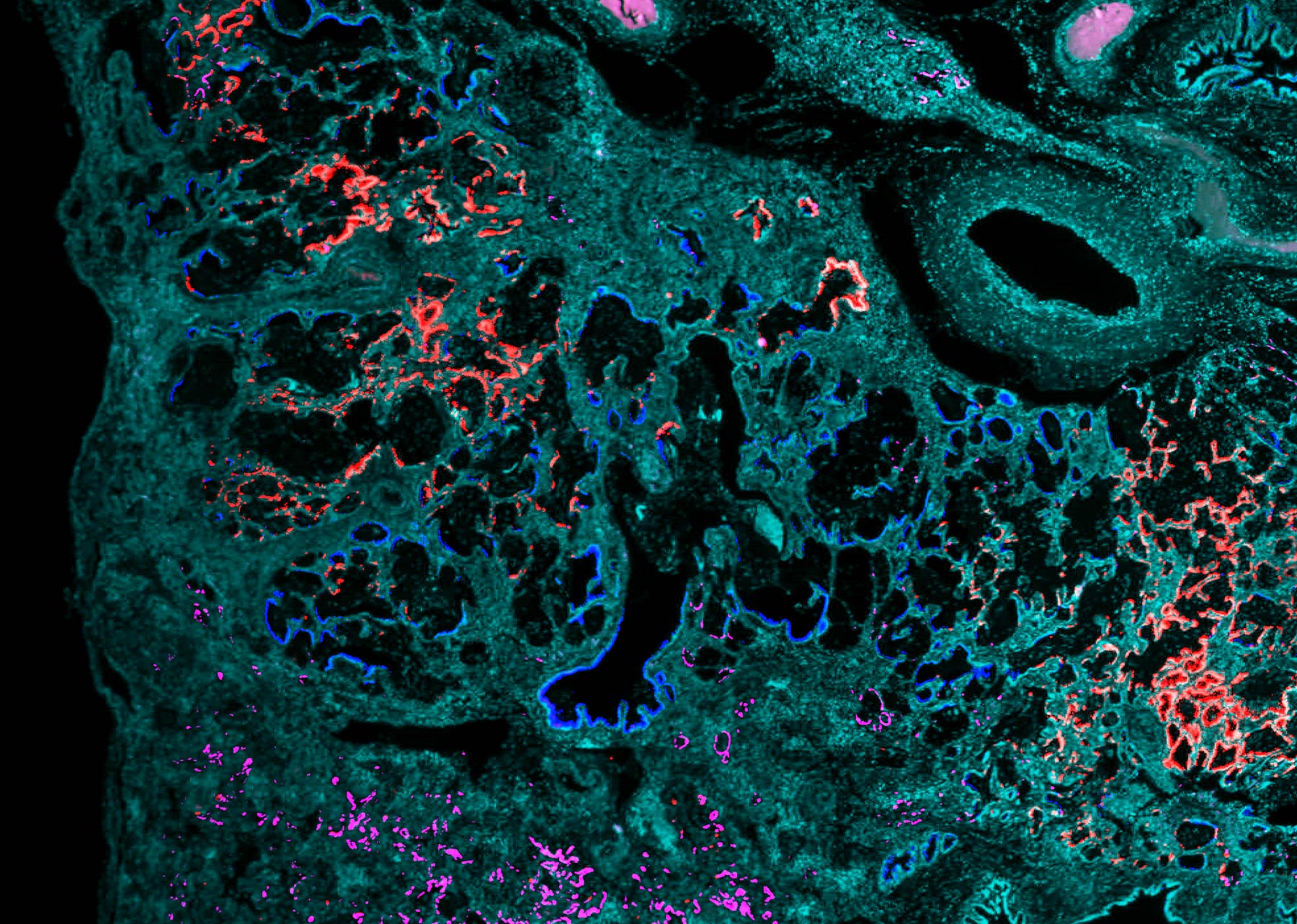
In-situ hybridization highlighting different secretory cell types in IPF lungs. Learn more about this IPF research here.
In 2020, Vanderbilt investigators published a seminal manuscript in which they (along with an independent group from Yale and Brigham & Women’s Hospital) used single-cell RNA-sequencing to identify numerous previously recognized cell types and states in IPF lungs, as seen in photo above. These findings reshaped understanding of the cellular organization of the human lung, and follow-up studies from Vanderbilt investigators and others have yielded new insights into key regulatory mechanisms that control epithelial cell plasticity in the context of lung injury, repair and fibrosis (disease-interacting single-cell expression quantitative trait loci, activation of transcriptional regulators including YAP/TAZ and HIF, as seen in photo below), immunopathogenic disease drivers and characterized the spatial evolution of fibrotic remodeling.
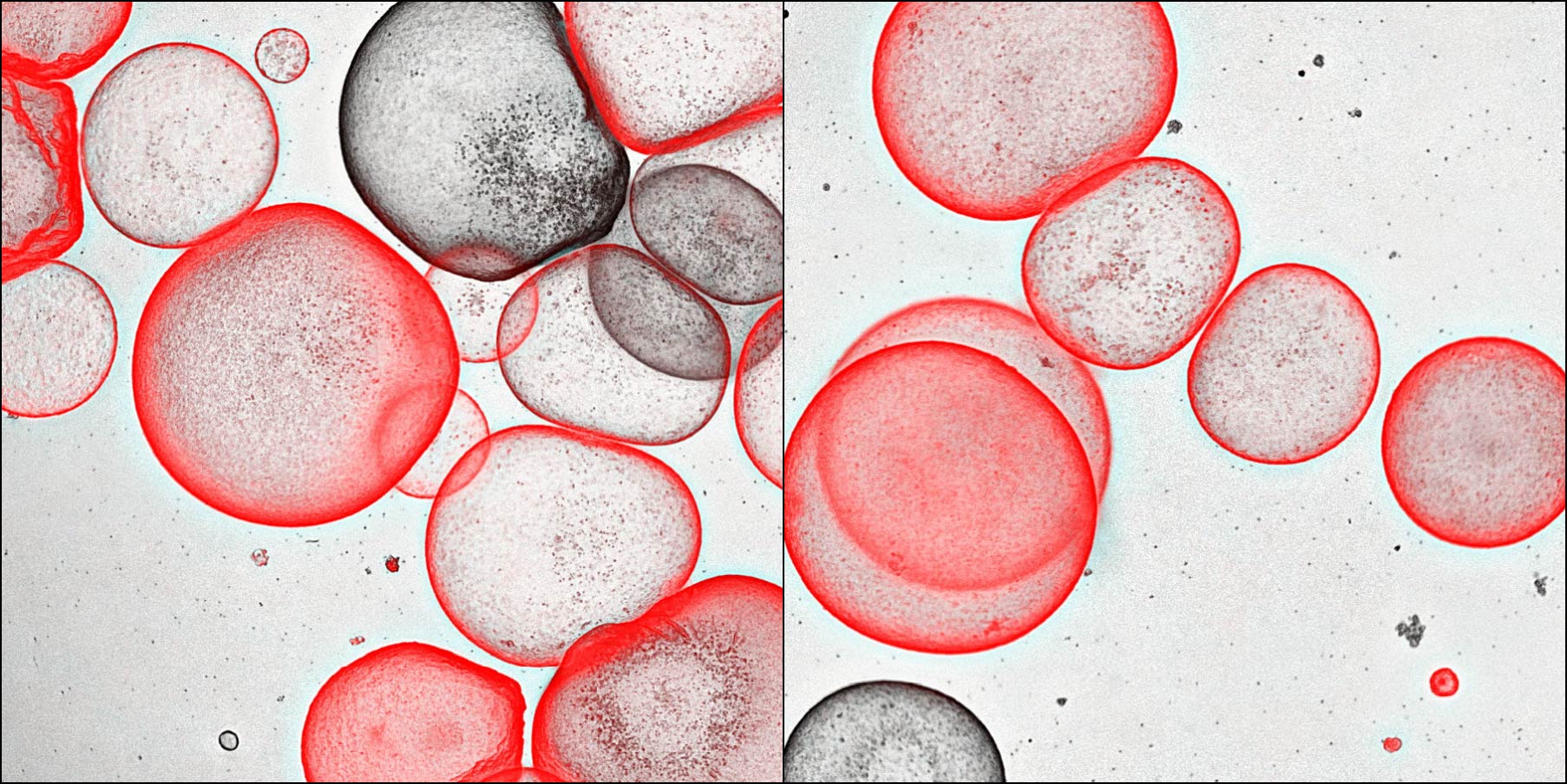
Alveolar organoids can be generated from distal airway secretory cells. Learn more about this HIF research here.