Pulmonary Fibrosis Research Studies
1. Family studies of IPF patients: Genetic and environmental risk factors for pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive fatal lung disease of unknown cause, with limited available therapies. Our clinical experience suggests that IPF has a familial or genetic basis and that relatives of patients with IPF have a 6- to 100-fold increased risk to develop pulmonary fibrosis, known as Familial Pulmonary Fibrosis (FPF).

Vanderbilt IPF/FPF registry
Since the Pulmonary Fibrosis Research Group established The Vanderbilt IPF Registry in 1999, we have enrolled more than 750 families of patients with IPF or other interstitial lung disease, as well as asymptomatic, first-degree relatives of IPF patients. Participants are asked to provide medical and family genealogy information, give a blood sample for isolation of DNA, and complete a survey about different exposures that may be risk factors for pulmonary fibrosis.
One of the fundamental objectives of this study is to identify genes that predispose individuals to develop IPF and understand the underlying mechanisms that lead to progressive lung fibrosis. Our goal is to use this knowledge to develop more effective strategies for treatment and prevention of this debilitating disease.
Environmental exposures
Another key focus of our research is geared towards identifying links between environmental exposures and pulmonary fibrosis. While there is growing evidence that numerous diseases are associated with environmental contaminants, there is limited understanding of how environmental exposures contribute to development and progression of pulmonary fibrosis.
To address this, asymptomatic relatives of FPF patients (termed “At-Risk for FPF”) in our FIP Registry can also participate in our “Environmental Exposures in Pre-Clinical FPF” sub-study. This study utilizes state-of-the-art technology and wearable devices to determine specific environmental exposures associated with pre-clinical pulmonary fibrosis and understand the mechanisms through which these exposures lead to the development of PF. With this study, we hope to develop novel environmental interventions that may prevent or delay the onset of symptomatic disease.
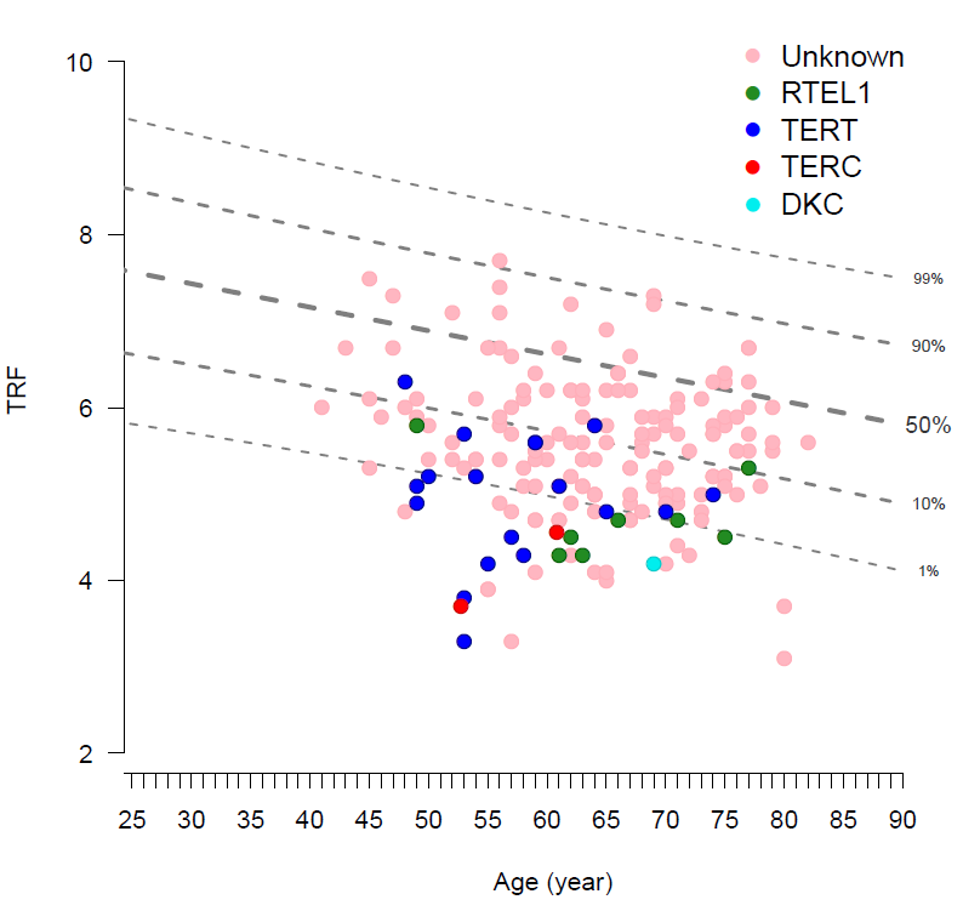
Short telomeres in peripheral blood are commonly found in patients with Familial Pulmonary Fibrosis.
2. Mechanisms of familial pulmonary fibrosis
Because family history is a major risk factor for pulmonary fibrosis, we established the “At-Risk for FPF” Cohort in 2008 to study the pre-clinical natural history among asymptomatic relatives of FPF patients. This cohort presents a unique opportunity to identify critical mechanisms driving the pathobiology of FPF by focusing primarily on the pre-symptomatic period, which represents the ideal time for interventions to modify the course of pulmonary fibrosis.
To date, there are more than 500 participants enrolled in this study, who undergo serial screening for pre-clinical PF with high-resolution CTs, pulmonary function tests, and blood donation. A key sub-study nested in this cohort includes the “Lung Sampling (Bronchoscopy) in Pre-Clinical FPF,” in which participants undergo lung sampling via a research bronchoscopy with bronchoalveolar lavage (BAL), bronchial brushing to collect airway epithelial cells, and transbronchial lung biopsies.
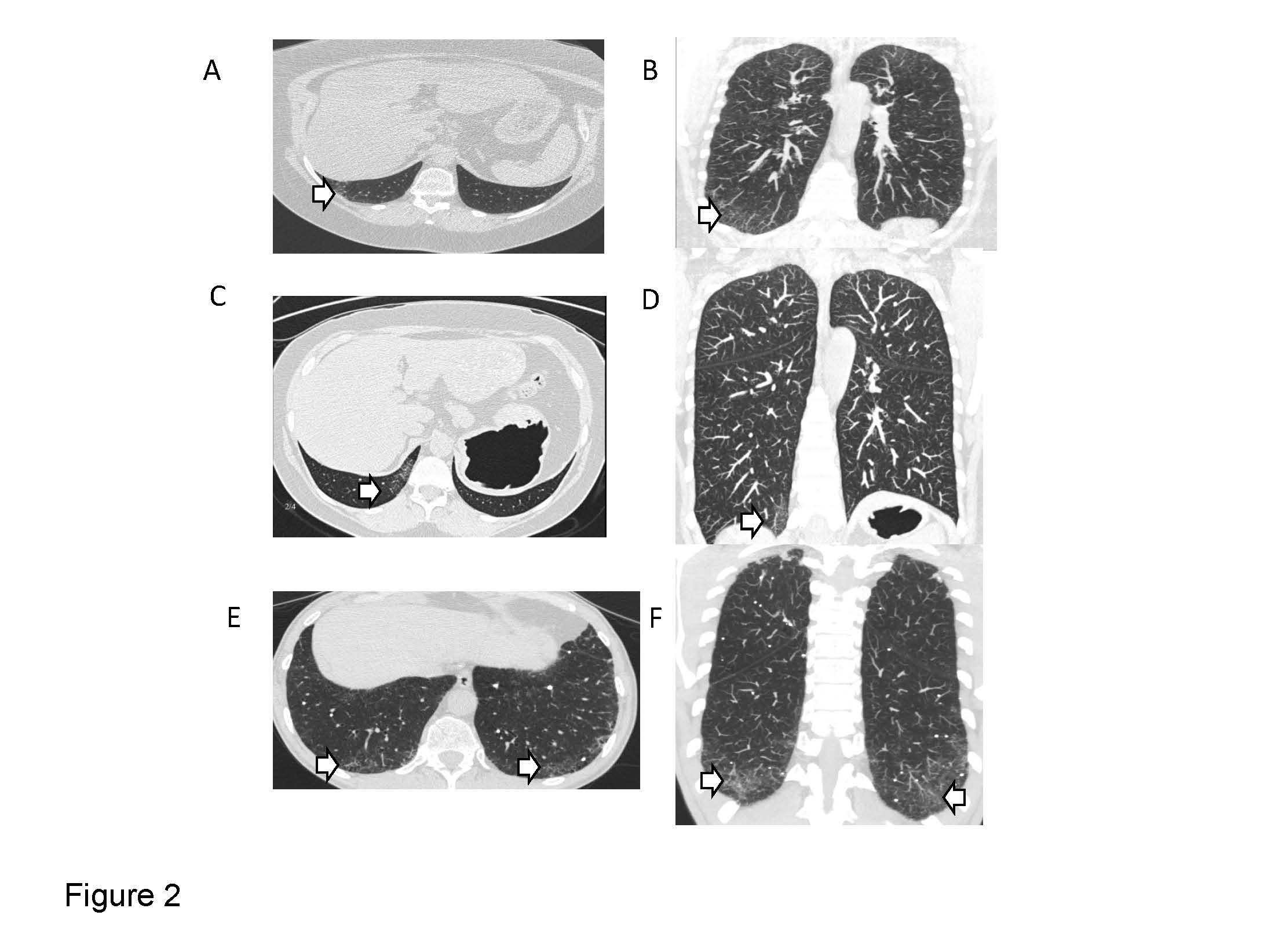
Early signs of pulmonary fibrosis (interstitial lung abnormalities) can be identified in FPF family members. Learn more about this research.
3. Pulmonary fibrosis at single cell resolution
The overarching goal of this research program is to investigate the underlying cellular and molecular mechanisms that drive disease initiation and progression of pulmonary fibrosis.
To achieve this, our research group utilizes a rich repository of biospecimens collected from human subjects in different stages across the pulmonary fibrosis spectrum and leverages advanced, state-of-the-art omics technologies to study the transcriptomic, genetic epigenetic, metabolic and exposome-related signatures that characterize pulmonary fibrosis across its natural history.
Our long-term objective is to use the knowledge gained in this program to develop new strategies for pre-clinical risk stratification, identify new therapeutic targets and develop novel approaches for disease prevention and treatment.
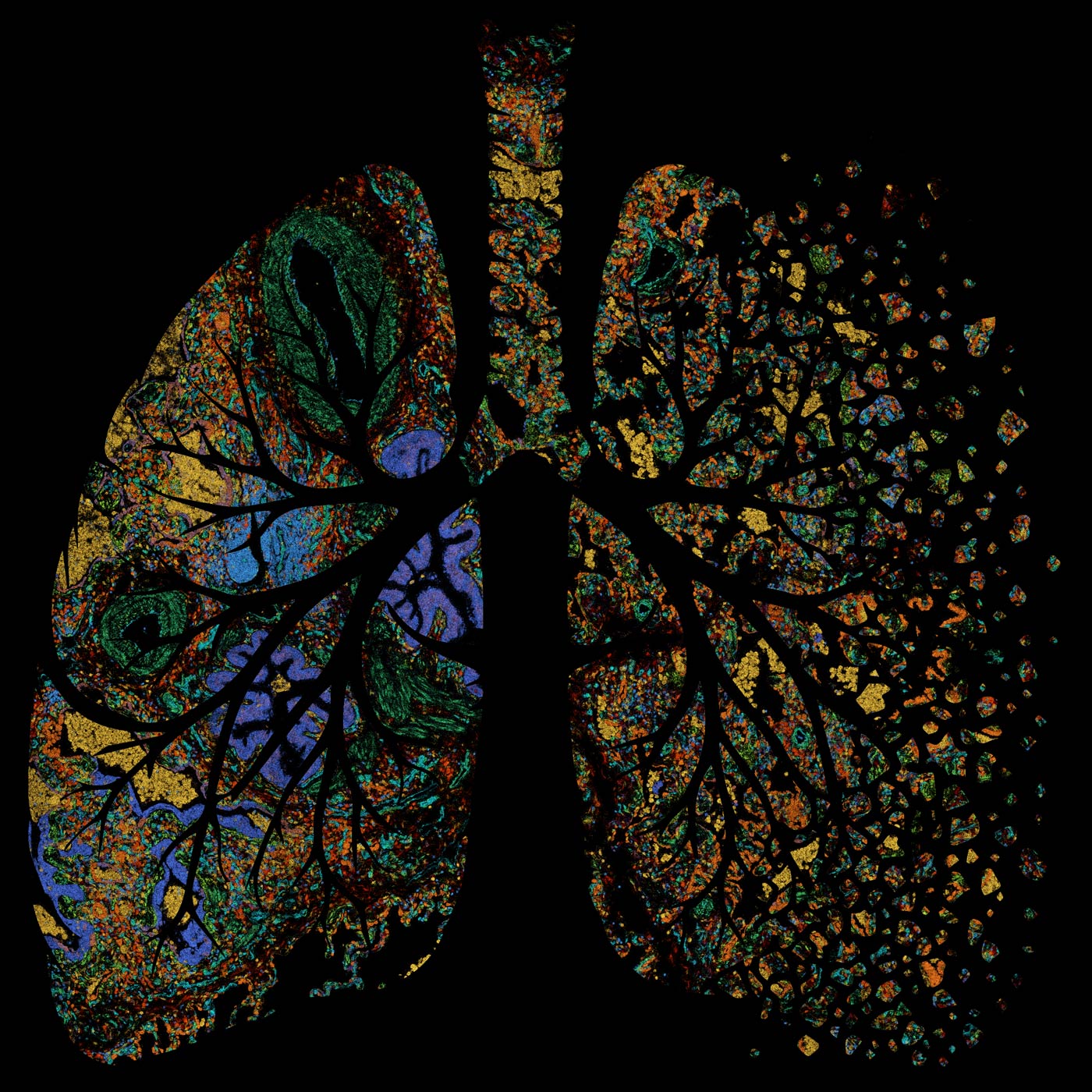
Spatial transcriptomic characterization of pulmonary fibrosis. Learn more about this research.
4. Immunopathogenesis of interstitial lung disease
Immune/autoimmune mechanisms are a frequent cause of interstitial lung disease, yet the mechanisms through which adaptive immune dysregulation drives lung fibrosis is not yet well understood.
Using state-of-the-art multiomic/single cell approaches, ongoing studies focus on autoimmune B-cells in myositis-spectrum interstitial lung disease and the role of tissue-resident T-cells in pulmonary fibrosis, and how immune aging impacts the development of interstitial lung disease.
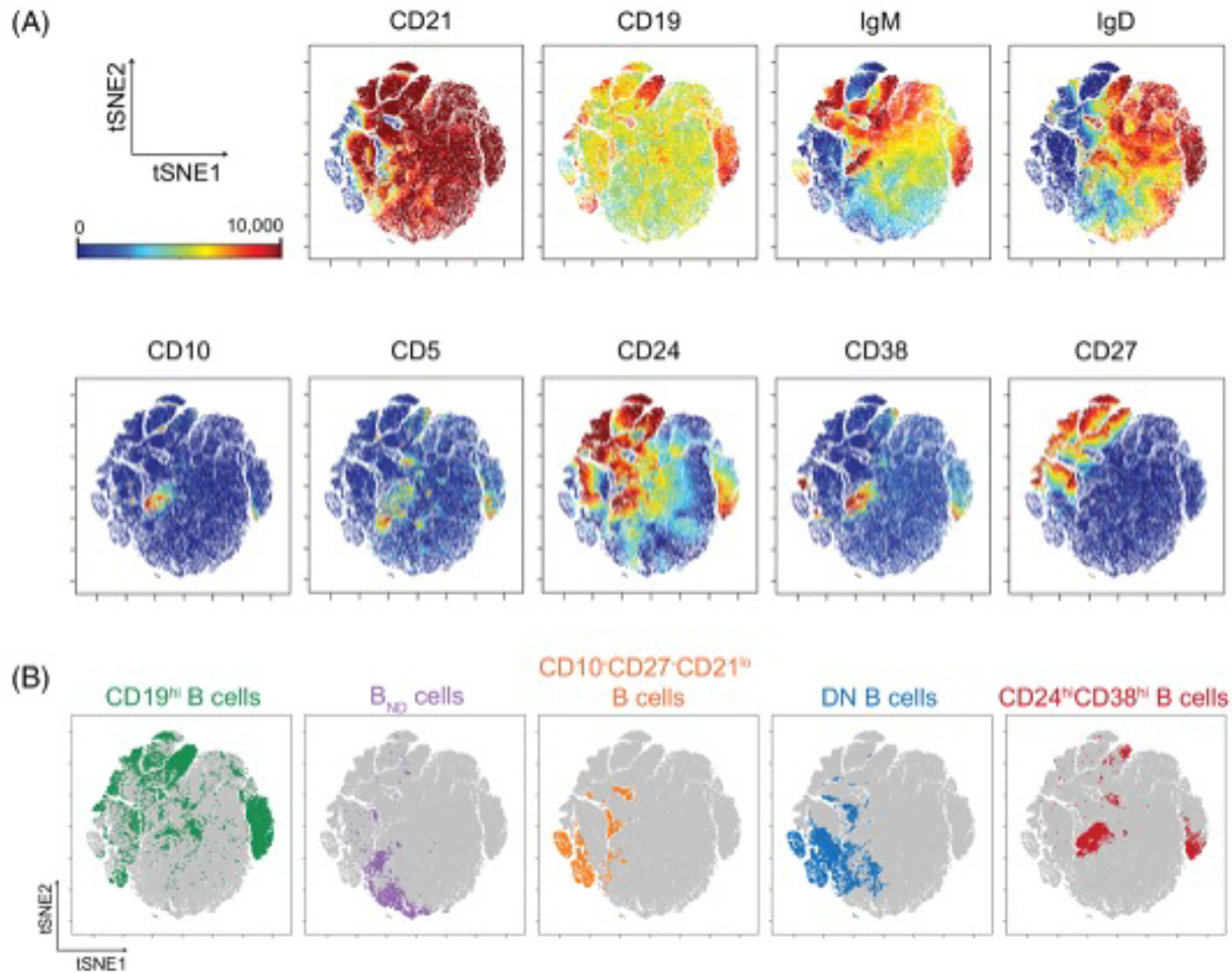
vISNE plot highlighting autoimmune cell populations from the blood of patients with autoimmune diseases.
5. Mechanisms of lung epithelial development, homeostasis and repair
Dysfunctional repair of the injured lung epithelium is a key feature of pulmonary fibrosis, and genetic evidence has suggested that this process is key in initiating the disease. Ongoing work in our group focuses on cellular signaling and transcriptional regulation of lung epithelial fate decisions during development, homeostasis and repair.
Using transgenic animal models, mouse and human ex-vivo models, ongoing studies are investigating the role of the orphan G-protein coupled receptor GPR87 as a regulator of epithelial repair, the influence of Hippo-effector transcription factors YAP and TAZ in alveolar epithelial repair, how the hypoxia-inducible factors (HIFs) control metabolic processes that govern alveolar epithelial cell function, and how inflammatory signaling through NFkB directs lung epithelial injury-responses.

